Seamless Cloning Kit (无缝克隆试剂盒)(D7010S) |
您所在的位置:网站首页 › 翻译 seamless › Seamless Cloning Kit (无缝克隆试剂盒)(D7010S) |
Seamless Cloning Kit (无缝克隆试剂盒)(D7010S)
|
使用说明: 1. 线性化载体的制备 载体的线性化可以在目标载体上选择合适的位点进行双酶切或单酶切,并对酶切后的载体进行琼脂糖凝胶电泳和切胶纯化(例如碧云天的D0056, DNA凝胶回收试剂盒)。也可通过在质粒插入位点的左右两侧设计方向相反的引物进行PCR,以获取线性化载体。PCR扩增获得的线性化载体,也需要琼脂糖凝胶电泳和切胶纯化。同时需要注意如下事项: a. 为了降低载体自连背景,提高阳性率,建议采用双酶切的方法对载体进行线性化。载体单酶切容易造成载体切割不完全和自连,导致假阳性的产生。 b. 酶切后无论是5' 端突出、3' 端突出还是平末端都可以用于本试剂盒,但由于重组过程中会产生3' 端突出末端,因此酶切后 形成3' 端突出或平末端的假阳性率会更低一些。 c. 通过酶切获得线性化载体的情况,线性化载体上的重叠序列只能用酶切后的3' 端序列进行计算。特别是对于酶切后产生5' 端突出的情况,不能以5' 端突出部分计算重叠序列,因为重组反应时有DNA 5' 外切酶(5' Exonuclease)活性,会消化掉5' 端序列。 d. 酶切较长时间,例如酶切3小时以上或酶切过夜,可有效降低载体未切开或自连导致的背景。 e. 通过PCR方法获得线性化载体时,建议在PCR后使用DpnI (例如碧云天的D6257)进行消化,以消除模板质粒对于后续获取重组质粒的干扰。或者在PCR之前宜先通过双酶切进行线性化处理,以消除模板导致的假阳性克隆。 f. PCR扩增获取线性质粒时,宜使用高保真的DNA聚合酶,例如碧云天的BeyoFusion™ DNA Polymerase (D7220, D7221),BeyoFusion™ DNA Polymerase (D7222, D7222B)或Pfu DNA Polymerase (D7216, D7217)。 2. 待插入片段的制备 a. 如果待插入片段是非常小的片段,例如20-70bp,可以通过单链DNA(常被称为引物)的合成和退火进行制备;如果待插入片段的长度比较长不能直接进行合成,可以通过PCR扩增的方法进行制备。 b. 无论是合成退火还是PCR扩增获得的待插入片段,在其两侧必须含有15-25bp序列与拟连接的载体或DNA片段完全重叠,后续重叠区域会发生重组,从而获得预期的重组质粒(参考图1)。 c. 通过PCR扩增获得待插入片段时,可以在设计引物时在两条引物的5' 端加上与拟连接的载体或DNA片段上完全重叠的15-25nt的序列。引物设计的要求和普通的引物设计要求相同,需要适当避免引物自身的二级结构、形成引物二聚体和GC含量过高等;并且在引物的5' 端加上15-25nt后,仍然要求整条序列适当避免自身的二级结构、二聚体和GC含量过高等。 d. 插入单个片段的引物设计方法可以参考图3。图3A中给出了常规的无缝克隆的引物设计方法实例的示意图,获得的克隆完全是无缝的。图3B中给出了保留插入位点两侧酶切位点的引物设计方法实例的示意图,获得的重组克隆可以保留两侧的酶切位点,以便于后续的酶切鉴定或者亚克隆等。 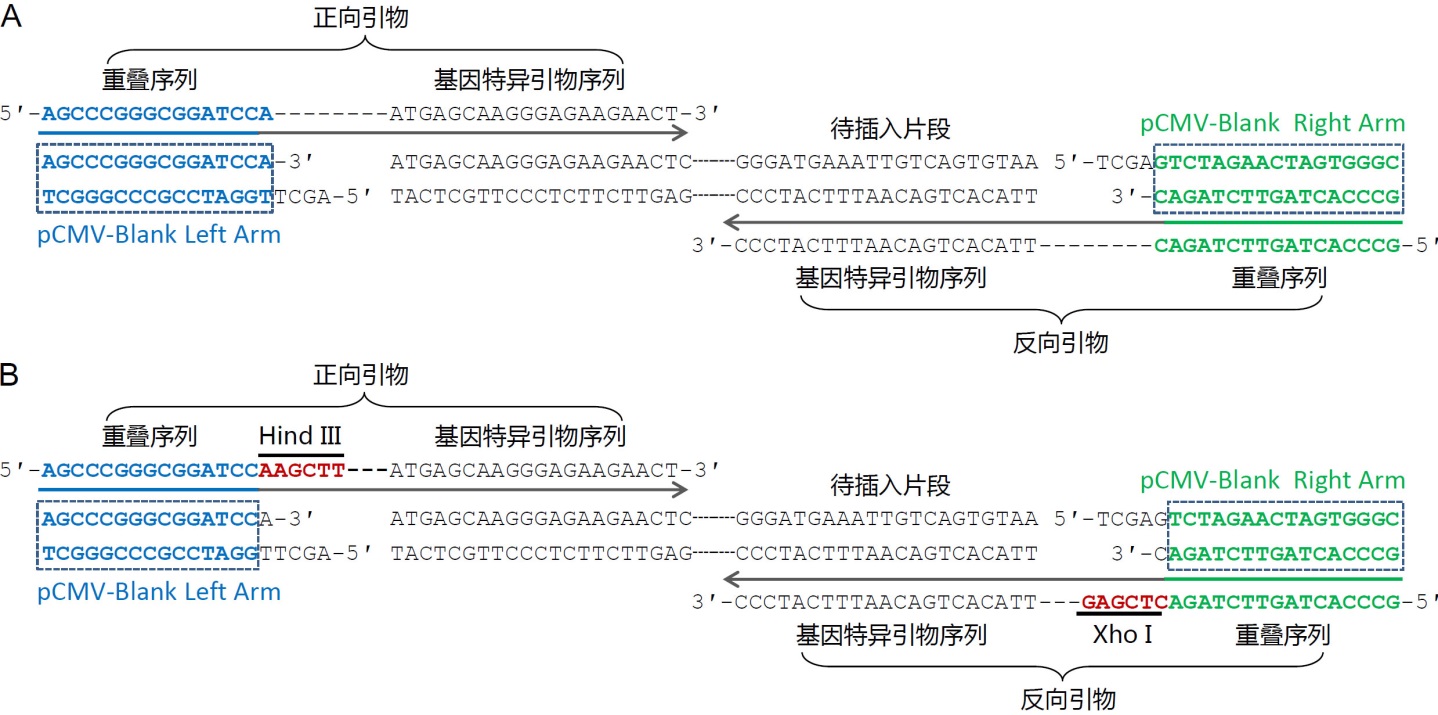
图3. 使用本试剂盒把单个片段插入到载体中的引物设计实例示意图。A. 目的序列插入到pCMV-Blank (D2602)载体中的无缝克隆引物设计示意图。pCMV-Blank用HindIII和XhoI双酶切线性化。选取图中所示的包含了重叠序列和基因特异引物序列的引物用于PCR扩增待插入片段,PCR产物切胶回收后与线性化的pCMV-Blank使用本试剂盒进行重组反应。B.目的序列插入到pCMV-Blank载体中并保留酶切位点的引物设计示意图。pCMV-Blank用HindIII和XhoI双酶切线性化。选取图中所示的包含了重叠序列、酶切位点和基因特异引物序列的引物用于PCR扩增待插入片段,PCR产物切胶回收后与线性化的pCMV-Blank使用本试剂盒进行重组反应。前者的优点在于引物设计简单,可以实现无缝克隆,后者的优点在于多了两个酶切位点,便于后续的酶切鉴定或亚克隆。此外,如果计划使用本试剂盒把目的片段克隆到碧云天的pCMV系列质粒中,上图中的重叠序列可以参考使用。 e. 插入多个片段的引物设计方法可以参考图4。通常可以先列出所需克隆片段的序列,标注出片段之间的相邻处,从相邻处挑选15-20bp作为重叠序列。重叠序列的挑选需要满足常规的引物设计要求,即需要适当避免引物自身的二级结构、形成引物二聚体和GC含量过高等。重叠序列可以仅在一个插入片段中(图4中红色DNA序列),也可以分布在两个片段中(下图中蓝色DNA序列)。重叠序列宜尽量设计为8个G或C和8个A或T,同时使退火温度不低于48℃(以AT配对为2℃,GC配对为4℃进行粗略计算)。随后利用重叠序列和基因特异的序列来设计相应的PCR引物,每个片段单独扩增后,所获得的PCR产物经琼脂糖凝胶电泳和切胶纯化即可用于本试剂盒的重组反应。 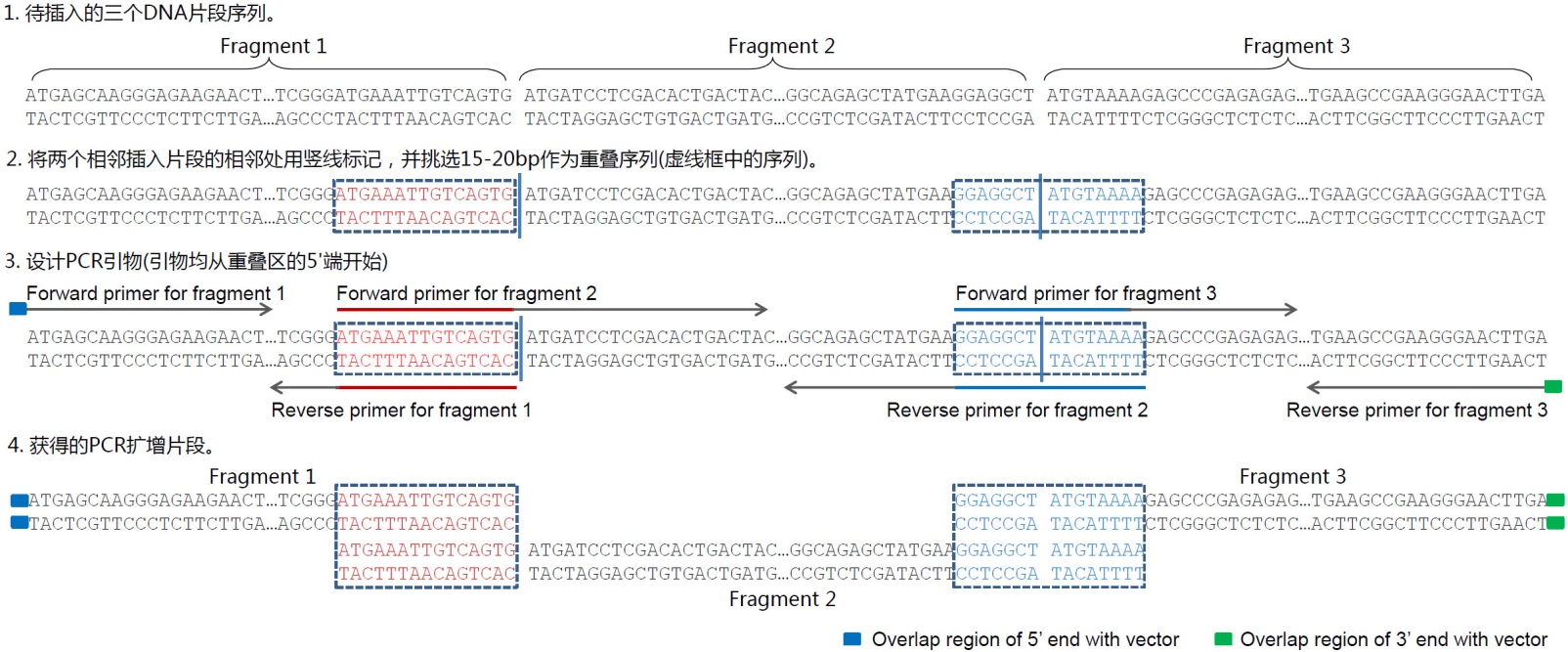
图4. 使用本试剂盒把3个片段插入到线性化载体中的引物设计实例示意图。如果需要插入更多的片段,可以参考上述实例示意图进行。 3. 取出试剂盒的4个组分,Nuclease free water置于室温,其余置于冰上。参考下表配制如下反应体系: 插入片段 1-2个片段 3-5个片段 阴性对照 阳性对照 每个插入片段与载体的摩尔比 3:1 1:1 - - 纯化的PCR片段 10-100ng 10-100ng - 2μl DNA fragment (1100bp, 30ng/μl) 线性化载体 50-100ng 50-100ng 50-100ng 2μl Linearized pUC18 (25ng/μl) 2X Seamless Cloning Mix 10μl 10μl 10μl 10μl Nuclease free water ? ? ? 6μl 总体积 20μl 20μl 20μl 20μla. 当插入1-2个片段时,片段和载体的摩尔比为3:1左右时,重组效率最高。 b. 当插入片段小于200bp时,建议插入片段和载体的摩尔比为5:1。 4. 上述反应体系置于50℃孵育15 min(插入1-2个片段)、30 min(同时插入3个片段)或60 min(同时插入4-5个片段)。反应完成后如不能立即进行后续操作,可将反应样品保存于-20℃。 5. 转化及克隆鉴定。 a. 取5μl上一步骤的反应样品加入到50-100μl DH5α等感受态细胞中(注意所加入的DNA样品体积不要超过感受态体积的1/10),轻轻混匀,将该混合物置于冰上30min。 b. 42℃水浴中90秒进行热激,然后迅速放回冰浴中,静置3~5min。 c. 加入500 μl不含抗生素的SOC或LB培养液,轻轻混匀,37℃震荡培养1h。 d. 将菌液5000 rpm离心1 min以沉淀菌体。吸去大部分培养液,剩余约50-100μl的培养液,重悬菌体。然后全部均匀涂布到含 适当抗生素的LB平板上,37℃培养箱中培养过夜。 e. 第二天挑取平板上的克隆进行菌落PCR或者提取质粒进行酶切鉴定,也可以直接选取数个克隆进行测序鉴定。 常见问题: 1. 转化效率低,菌落很少甚至没有 a. 为确保试剂盒正常工作,请用试剂盒提供的线性化载体和插入片段进行对照实验。 b. 引物序列设计不正确。检查引物序列,确保引物两端有15-25bp的同源臂以用于与相邻片段进行同源组装。 c. 感受态细胞对重组反应体系过敏。使用水对重组后的反应液稀释5-10倍后再进行转化。 d. PCR产物不纯或插入片段即线性化载体浓度过低。采用凝胶电泳和胶回收的方法对PCR产物和线性化载体进行纯化,严格按照说明书推荐的摩尔比和浓度进行插入片段和线性化载体的混合。在20μl总反应体系中,插入片段和线性化载体混合后体积应尽量小于5μl,但如果插入片段和线性化载体用水溶解或洗脱的,体积可以不受此限制。 e. 转化细菌时重组反应样品加入过多。确保重组反应样品的体积不超过感受态细胞体积的10%。 f. 感受态细胞的转化效率过低。用高效率感受态重新转化。 g. 选择了错误的抗生素,或在固体LB培养基中加入了过量抗生素。涂板时选择正确的抗生素和浓度。 2. 阳性率过低 a. 载体线性化不完全。建议延长酶切时间至过夜并凝胶电泳后切胶回收。 b. 模板质粒污染。以质粒为模版进行PCR扩增,得到的PCR产物最好经过凝胶电泳和切胶纯化,这样可以有效去除模板质粒。使用载体为模板通过PCR扩增得到线性化载体时,推荐使用DpnI酶消化以消除模板质粒的干扰。 c. LB平板过期或用平板配制时使用了错误的抗生素,确保使用的LB平板是1个月内配制的,并检查载体的抗性。 3. 阳性克隆含有错误的插入片段 PCR产物含有不正确的插入片段。优化PCR反应引物设计以增加PCR产物的特异性,并对PCR产物进行凝胶电泳和切胶纯化。 4. 平板上有很多小菌落 有些外源蛋白的表达对大肠杆菌是有毒的,此时需要考虑采用低拷贝质粒进行克隆。 |
【本文地址】
今日新闻 |
推荐新闻 |